《2022 Artificial Intelligence in Drug Design》 笔记--第二章:机器学习在药学和ADMET终点建模中的应用
近年来,定量构效关系(QSAR)这一广为人知的概念引起了人们极大的兴趣。数据、描述符和算法是构建有用的模型的主要支柱,这些模型支持使用计算机方法进行更有效的药物发现过程。这三个领域的重大进展是人们对这些模型重新产生兴趣的原因。在这本书的章节中,我们回顾了各种机器学习(ML)方法,这些方法利用了许多化合物的体外/体内测量数据。我们将这些与其他数字药物发现方法联系起来,并给出了一些应用实例。近年来,定
目录
第二章:机器学习在药学和ADMET终点建模中的应用
摘要
近年来,定量构效关系(QSAR)这一广为人知的概念引起了人们极大的兴趣。数据、描述符和算法是构建有用的模型的主要支柱,这些模型支持使用计算机方法进行更有效的药物发现过程。这三个领域的重大进展是人们对这些模型重新产生兴趣的原因。在这本书的章节中,我们回顾了各种机器学习(ML)方法,这些方法利用了许多化合物的体外/体内测量数据。我们将这些与其他数字药物发现方法联系起来,并给出了一些应用实例。
Key words: Artificial intelligence (AI), Machine learning (ML), Deep neural network, In silico
ADMET, Quantitative structure–activity relationship (QSAR), Pharmacological endpoint, Data sci-
ence, FAIRification, Physicochemical properties
1.引言
药物在主要靶点上的药理活性以及吸收、分布、代谢、排泄和毒性(ADMET)是新药发现和优化的主要参数。
这一点早已得到承认,制药/生物技术公司和学术机构已投入巨资开发新的分析方法,并提高其测试能力,使它们能够在高质量的分析中表征数百万种化合物。存储的结构-活性/结构-性质关系(SAR/SPR)数据可能是一个宝库,有可能影响这些测量所针对的特定项目以外的研究。计算小组一直在使用这些数据在Silico中寻找新的铅结构,对某些admet端点潜在的原理产生理解,并在Silico模型中开发可作为辅助工具的附加工具研究人员在优化导致药物[1]。这些模型的主要目标不是减少体外或体内ADMET实验的总数,而是让科学家更好地将他们的实验集中在最有希望的化合物上。
在这本书的章节中,**我们概述了在早期药物发现阶段应用于建模与决策相关的化合物属性的先决条件和计算方法,并与拜耳在过去20年中产生的硅胶自适应方法的发展进行了补充。**我们将专注于ADMET的特性,而不包括蛋白质化学计量学[2]、与靶标和靶外相互作用有关的靶标分析[3-5],以及多元药理学[6-8],即识别、设计或使用作用于多个靶标或疾病途径的药剂。
我们也不会讨论概念上不同的基于蛋白质结构的方法,在这些方法中,化合物与定义的蛋白质的相互作用被建模并用于设计更好的化合物,这对ADMET的性质非常重要。这种方法需要与单个ADMET相关蛋白(如细胞色素P450酶、PXR、HERG、PGP、HSA)明显相关的ADMET效应和这些蛋白质的高分辨率X射线结构。讨论了这些方法的应用和效用[9,10]。最近在冷冻电子显微镜方面取得的成功,例如在整个HERG通道上取得的成功[11],将对该领域产生重大的积极影响。
在这篇综述中,我们专注于第二种概念性方法,其中许多化合物的体外/体内测量数据被用来使用机器学习(ML)来建立模型。我们总结了相关的回顾、研究和拜耳在机器学习方面的经验,并将讨论定制的分子和原子描述符和算法的最新发展,如(深度)神经网络。最后,我们提供了精选的应用实例,特别强调整体药物发现方法。
基于结构的方法和基于机器学习的方法都可以应用基于经验或基于物理的对分子及其相互作用的描述。经验函数的例子是对接分数和我们在下一节中描述的大多数分子的数值描述符。
另一方面,基于物理的方法使用物理定律来描述分子系统。在量子力学中,分子体系完全由其电子结构决定。通过求解用电波函数描述系统的薛定谔方程,可以获得所研究系统的能量。由于薛定谔方程的精确解只对最小系统是可行的,所以出现了精确公式的近似形式。近似的类型和范围与计算的成本,在不同的质量管理方法之间有所不同。到目前为止,工业上最常用的质量管理方法是密度泛函理论(DFT),这主要是因为它具有有利的成本-精度比。
有几种性质,如化学反应活性、键能或电子光谱,只有通过使用QM才能准确捕获。在原子描述符的一节中,我们将提供来自QM计算的分子抽象的例子。
在分子力学中,分子系统是用粒子(如原子)的位置和动量来描述的。系统的势能可以由两两势能给出,也称为力场,决定了所有键合和非键合的相互作用。通过求解牛顿运动方程,可以像在分子动力学(MD)模拟中那样研究分子系统的动态演化。给出构象空间的充分采样和自由能估计器的使用,例如多态Bennett接受率(MBAR)[13],可以从MD模拟数据(更多信息见,例如参考文献)导出用于描述例如蛋白质-配体结合的自由能差。14-16)。自动化、方法论和计算能力方面的重大进步使分子动力学和精确的自由能计算在工业中得到越来越多的采用[17]。在一个应用程序示例中,我们将展示如何利用这种方法。
2.机器学习在ADMET问题中的应用
2.1良好的ADMET性质的重要性
从上世纪九十年代开始,甚至在本世纪初,很明显,仅有靶向亲和力和选择性不足以将药物推向市场。有利的物理化学和药代动力学参数具有特别重要的意义,因为到目前为止,全球市场上所有剂型中约有80%是口服给药,因为这是患者最方便的形式[18]。然而,其他给药途径也对药物的性质提出了特殊要求,例如在静脉给药的情况下具有非常高的溶解度。
在2000年前后,越来越清楚的是,后期的磨损与较差的化合物性能直接相关。Kola和Landis在2004[19]中指出,在1991到2000年间,由于PK和生物利用度的原因导致的磨损率显著下降(从42%下降到10%),而在同一时期由于毒理学和临床安全性而导致的磨损率显著增加,这归因于化合物的尺寸和亲脂性的增加。这些发现后来被阿斯利康、葛兰素史克、辉瑞和礼来联合发表的一篇论文所证实[20]。
确定决定候选药物风险因素的物理化学参数的最早尝试之一是利平斯基对五规则的开创性工作[21]。像文洛克[22]、利森[23]和格里森[24]这样的几位研究人员进一步分析了特性曲线及其与体外效力和ADMET的联系。这导致了可供选择的规则集,如Veber规则[25],格里森的“可解释的admet经验规则”[26],“金三角”[27],以及铅类化合物的三规则[28]。
ADMET机器学习是从定量结构-活性或结构-性质关系(QSAR/QSPR)领域发展而来的,它起源于20世纪70年代[29],始于线性相关模型。这一方法的广泛应用体现在2003年建立的包含18,000个模型的C-QSAR数据库[30]以及最近的审查,如“QSAR无边界”[31]和“化学信息学和药物发现中的机器学习”[32]。
通过分析谷歌趋势报告的互联网搜索频率和谷歌学者报告的出版物数量,可以说明该领域的重要性。
谷歌趋势(https://trends.google.com)通过与客户执行的查询总量相比较的频率来衡量特定术语和关键词在一段时间内的受欢迎程度。对2004年1月至2020年10月期间的“QSAR”、“机器学习”、“深度学习”和“admet”四个词的设置为“全球”和“所有类别”进行了趋势分析,并按月进行解析。总体上最重要的术语的趋势得分被设置为100,而如果该术语没有显著地比所有索引术语的平均值更相关的情况下的最低得分被设置为零。
每年汇总的数据如图1a,b所示。与“机器学习”相比,“定量构效关系”一词并不十分突出,2004年的最高得分为4.8分,2012年降至约1分。术语“admet”一直是一个专业术语,得分始终在1左右。另一方面,由于这个术语的普遍使用,机器学习在2015年的重要性上升到2019年的高分,得分从10到20分,达到91分(12个月的平均值)。
深度学习作为机器学习中的一个子学科也在2015年左右开始兴起,但它的最高得分在40分左右。尽管趋势分析具有启发性,但人们必须记住,它始终只是一个相对的衡量标准。例如,将“机器学习”与可能相当流行的术语“气候”进行比较,2004年至2020年期间的平均分数分别为12分和42分,2020年的平均分数为56分和68分。得分的相对性甚至通过比较2020年的术语“机器学习”和“宝马”得到了更明显的说明,这两个术语的得分分别为1和83,说明了科学术语和普遍感兴趣的术语之间的差异。
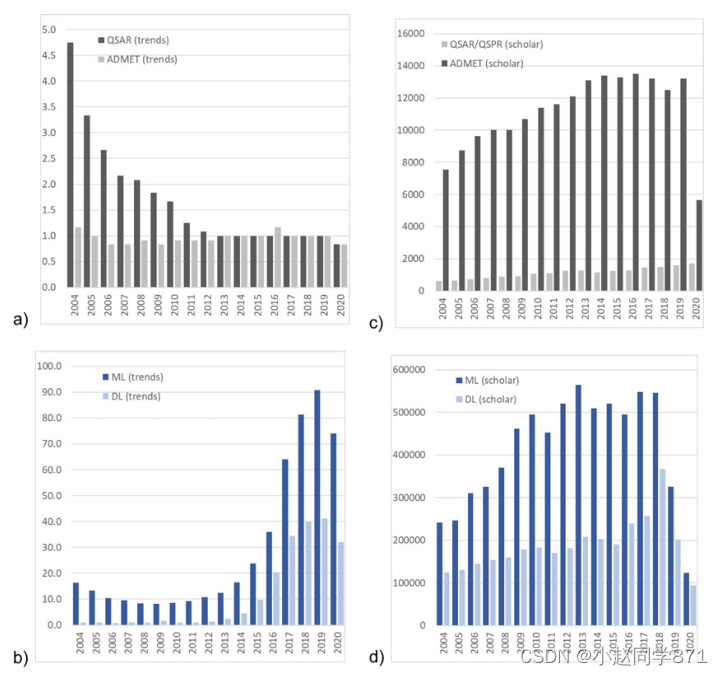
插图。谷歌趋势分析(A)QSAR、ADMET和(B)机器学习(ML)和深度学习(DL)在2004至2020年间的出现情况,包括设置为“全球”、“所有类别”、“网络搜索”和谷歌学者©和(D)。详情请参阅正文
第二个分析是根据谷歌学者(https://scholar.)的数据,包含特定术语的出版物数量。设置为“任何语言”,但不考虑“专利”和“引文”。报告的数字是估计数字,而不是谷歌所说的确切数字,但仍然可以确定总体趋势。
随着时间的推移,带有“QSAR QSPR”一词的出版物数量增加了三倍,从620种增加到1700种(见图1)(可能与多年来出版物的总体增长同步)。其他期限的增幅只有两倍或更少。术语“admet”的数字要高出十倍,对于“机器学习”和“深度学习”,与“QSAR QSPR”相比,这两个因素分别约为400和200,后者的增长较晚,最近才在2017年左右开始。人们必须记住,在表演和研究发表之间有一段时间的延迟。2018年“气候”和“宝马”的搜索量分别为393,000次和18,200次,相比之下,“机器学习”的搜索量为547,000次。
我们看到,“机器学习”在科学和公众中是一个相关的话题,更具体的术语排名明显不那么重要,而且“QSAR”现在已经过时了。但与这个术语无关的是,数据驱动的模型在药物发现和任何其他学科中都具有高度的相关性。
2.2数据、描述符、算法、指标
任何机器学习模型都是一个或多个可观察数据端点之间的数学关联,即因变量,以及要建模的对象的描述,在我们的情况下,通过描述符来建模,在我们的情况下,描述符是自变量。虽然早期应用的算法是线性算法,如多元线性回归,但很快就变得明显的是,相互依赖实际上是非线性的,这导致了越来越复杂的算法的发展,如贝叶斯方法、支持向量机、随机森林、人工神经网络,仅举几例。已经证明,通过应用更复杂的方法在预测性方面的额外收益会从一个水平恶化到另一个水平[33]。
在下面,我们讨论稳定和预测模型的三个关键组成部分,即数据、描述符和算法,以及应用于识别这些模型的度量标准。在后面的部分中,我们将讨论模型更新的最佳实践流程和策略。
2.3数据是关键
任何机器学习模型的底层数据都将决定模型的质量和稳健性。这一事实在某种程度上被大量的出版物掩盖了,这些出版物大多是由学术机器学习社区发布的,这些出版物的中心是新的描述符和算法。有两种类型的数据,即学习的化学结构和分析数据。这两种类型的数据在应用于机器学习之前都需要一些数据整理步骤[34,35]。
2.3.1 实验分析数据
在制药行业,实验数据通常存储在公司数据库中,并可直接访问。但这并不意味着数据是开箱即用的机器学习准备。历史上,分析定义和上传过程通常以允许请求的研究项目直接使用数据的方式设置,但不考虑进一步的使用,例如分析参数通常是自由文本而不是受控词汇,重要信息存储在注释中,化验说明过于简短且不是不言而喻的。
2016年3月,一个科学家联盟发表了一份出版物[36],其中概述了四项基本原则–可发现性、可访问性、互操作性和可重用性–简写为公平原则,描述了FAIR过程的程序(https://www.go-fair.org/how-to-go-fair),,即清理历史数据和建立新的数据库的过程,着眼于未来。虽然最初是作为一项学术倡议,但这些原则现在也在行业中进行了调整,以提高数据质量,提高对历史和未来数据的访问,并允许从原本不相关、不完整和有噪音的数据中获得见解。
对于机器学习,必须提取用于兴趣分析的数据,并且必须排除模棱两可的结果。在这个过程中,与实验者的密切沟通对数据科学家来说是至关重要的,因为正如Brown等人[37]所描述的那样,多个实验参数决定了使用哪些数据以及从建模中排除哪些数据。
化验由四个部分组成:生物或物理化学测试系统、检测方法、技术基础设施,最后是数据分析和处理。在CaCO2渗透试验的情况下,生物系统可以是人的结肠细胞,在细胞色素P450抑制的情况下可以是与蛋白质的相互作用,或者在体内试验的情况下可以是测试动物。检测方法可以是用于分离、鉴定和定量测试物质的高效液相色谱(HPLC),或UV光谱。技术基础设施由许多部件组成,如玻璃器皿、塑料管和吸管,以及井板,由全自动机器人系统运行。数据分析最终意味着任何程序,例如从原始数据点确定IC50值的曲线拟合。
生物系统具有一定的可变性,如细胞活动和细胞计数的差异或蛋白质活动和质量的差异。样本以非常小的体积间隔从测试系统中获取,增加了数据的可变性。该检测方法具有一定的可变性,在紫外光的情况下,高度依赖于测试分子中是否存在合适的生色团。该检测方法定义了检测的下限和上限,产生了前缀为“<”或“r”>的值,即所谓的删除值。
在靶组织中的最大暴露还取决于物质的最大溶解度。测试物质以二甲基亚砜(DMSO)原液的形式储存,然后稀释,使最终的DMSO含量通常为1%。这仍然会导致细胞活性降低,并扰乱结果。
高度亲油的化合物对玻璃或塑料部件(如吸管)的粘附性降低了化合物与表观浓度相比,导致数值错误地过高。部分溶解和沉淀也会改变测定体系中的实际浓度。研究物质的纯度通常低于100%,结晶状态往往没有很好地定义。
对于大多数化合物,只进行一次测试的事实阻碍了错误测量和异常值的识别。总而言之,这导致模型的输入数据具有一定的可变性,从而决定了可实现的可预测性。布朗等人。[37]分析了来自亲和分析的65,000个pIC50的变异性,得出了0.3个对数单位的这种分布的中位误差(对应于IC50值的系数2),这是根据《检测指南手册》的良好检测的预期变异性[38]。
2.3.2 化学结构标准化
尽管已在公司或公共数据库中注册或由文件提供,但按原样使用化学结构仍存在潜在的陷阱。数据库盒式磁带或用于写入文件的软件将检测并修复明显的语法错误,但不会更多。Young等人[38]在六个公共和私人数据库上的一项研究给出了从0.1%到3.4%的错误率,并证明了包括错误分子会导致模型精度显著下降。
化学结构文件格式在涵盖化学的范围上有所不同。例如,微笑[39]不能编码“或”–立体化学或相互依赖的立体中枢集合。这可能是由前MDL,现在的Biovia(2002-2020达索系统)制定的SDF标准[39,40],或国际化学识别符INCHI[41]。众所周知,存在微笑表示法等“方言”,人们应该预料到不同软件之间的细微差异。因此,为了模型的训练和应用,整个管道应该保持不变。
结构标准化是一个多步骤的过程[42],根据手头的问题,可能会有细微的差异,但总体过程总是相同的。由于其目的是以一种能够计算分子描述符(将在后面详细讨论)的方式来标准化分子,因此只有可以通过这种特征来明确描述的分子的特征必须被保留。在电荷态、立体化学和互变构态的情况下,应用严格的规则来优化描述符中的信息内容。这里的最终目标是提出一个标准化协议,该协议不仅适用于模型训练,也适用于模型应用(图2)。
首先,盐和混合物被分开,保留最大的片段或应用盐的匹配模式的列表,产生恰好一个化学实体。然后应用过滤器来去除不需要的化学物质,如无机物或有机金属,不完整的结构。根据建模任务的不同,基于分子量或结构模式匹配的多肽或大环等大分子也必须被过滤掉。在确定药理活性物质的实验性质的情况下,可能有必要分离前药的剩余基团。
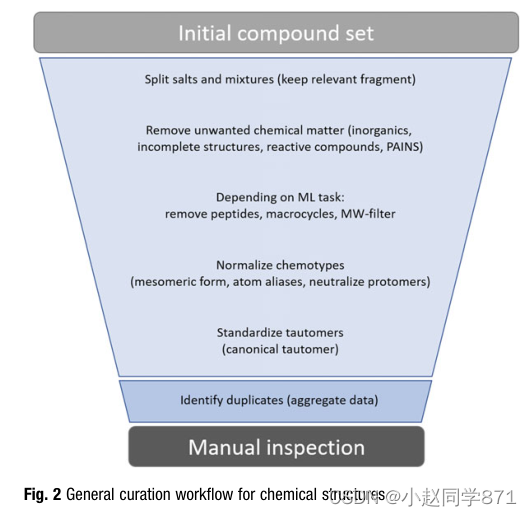
插图。化学结构的通用管理工作流程
存在于多个介体表示中的官能团,如芳香族和杂环族或例如硝基,将被归一化,并且原子别名(即,羧基或甲基的缩写为COOH或Me)必须被解析为显式原子。除了定义立体化学所需的显式氢原子外,化学结构通常存储时没有氢原子。不一致的氢处理可能导致不同的描述符值[42]。
分子的电荷状态和互变异构体形式取决于实验条件,如溶剂化作用或与生物目标的定向相互作用,这些条件很难确定。因此,只要有可能,酸性或碱性官能团被中和以获得全部中性配体(例如,存在像季胺这样的永久带电官能团)。
化合物可以以几种互变异构形式存在[43]。不同的靶蛋白具有不同的药理相关形式,并且依赖于溶剂。没有简单而明确的方法来计算相关的形式[44]。另一方面,对于数据集中的同种化合物,相同的易于互变构体的亚结构可能会以不同的方式表示,对于稍后要预测的化合物来说,甚至更有问题。一个可行的解决方案是严格标准化标准的互变异构体[45,46]。根据实施的规则集和实施的搜索策略,不同的软件工具将再次导致不同的规范互变异构体。有关全面基准的信息,请参考《范围》出版物[47]。
化合物不仅以不同的互变异构体和电荷状态存在,而且还以纯立体异构体和外消旋的形式存在。同样,由于软件之间的微小偏差,数据库和文件中提供的信息并不完全可信,还取决于用于立体化学阐明的应用分析过程的可靠性和实验室人员的正确报告。大多数描述符包无论如何都不能处理立体化学,因此立体中心是扁平化的。
最后,在对目标亲和力简档进行建模的情况下,可以在诸如Pain或“Hit Dexter”[48,49]之类的频繁点击者上另外应用结构过滤器,以避免由于非特定绑定数据而产生的噪声。
考虑到这一切,作为创新医学倡议(IMI,http://www.imi.europa.eu))的一部分,欧盟资助的联盟IMI MELLODDY(www.Mellowdy.eu)开发并发布了一个端到端的开源工具,用于MELLODDY_Tuner(http://www.mel lowdy.eu/开放源代码-代码库)所述的过程。该工具用于标准化项目成功所需的数据,以努力联合和保护隐私的机器学习利用世界上最大的具有已知生化或细胞活性的小分子集合,以实现更准确的预测模型,并提高药物发现的效率。
2.3.3机器学习测试数据的预处理
本节描述了对来自一个来源的实验数据的管理过程,如在公司建立和运行的分析通常需要很长一段时间。来自不同来源的数据的组合带来了更多的挑战,Kramer等人描述了从ChEMBL12提取的2540个蛋白质-配体对的7667亲和力值,其平均误差为0.44个PKI单位[50]。但即使是微妙的实验细节也可能导致实质性的不一致[51]。另一种选择是建立一个多任务ML模型(参见2.4机器学习算法),该模型预测一种分析的值并使用另一种变体作为辅助任务。
在最佳设置中,数据存在于实验水平上,即每个执行的实验都有一个结果,并且关于导出结果,如IC50或EC50是用于曲线拟合的所有测试点都是可用的,并且允许识别弱曲线拟合。
有三类数据需要管理:带有附加注释的数据、经过审查的数据,以及具有包括离群值在内的多个测试值的结构。附加在化合物上的注释,如“未完全溶解”或“校准问题”,允许过滤掉不可信的实验。删失数据是位于检测窗口或序列稀释窗口之外的数据。这类数据通常用前缀“>”表示为右审查数据,用“<”前缀表示低审查数据。如果化验的检测限发生变化,也可能会有中间审查的数据。对于分类器模型(稍后描述),可以使用此类数据,因为它们属于其中一个类。对于数值模型,必须排除它们,否则将应用专门的删失回归算法[52]。应该始终删除中间删减的值。由于删失值的实际实验值低于或高于给出的数值,因此在左删失数据和右删失数据的情况下,通常分别将其除以或乘以系数2。
多重值的处理是最复杂的话题,没有一刀切的解决方案。首先,它取决于所应用的化学结构聚集。基于特定批次的分子的多个值允许识别分析本身的可变性。如果有严重异常值的例子,化验原理本身可能会受到质疑。分子水平上聚集的异常值是批次可变性的暗示,即化合物纯度的可变性。或者,如果涉及多种盐型,则取决于盐型的影响。但如前所述,至少在大多数情况下,我们和其他人是基于无立体化学的最大片段聚集的。通过这一点,我们接受由于批次、盐和立体化学差异而产生的可变性。
在任何这样的聚合级别上,都必须处理离群值。去除是最严格的方法,需要一种复杂的策略,不同的检测方法会有所不同。通常,如果值与数据分布相差两到三个标准差,则被视为异常值,这对于值的数量较少是有问题的,对于经审查的数据也是一个问题。因此,对于非目标分析,如ADMET投资组合,我们决定应用风险最高的值,如在CYP或HERG抑制的情况下的最低值,或者其他情况下的中位数,它对异常值比平均值更稳健。
2.3.4数据保护的努力和重要性的示例
如前所述,Young等人的研究(s.o[53].)。报告称,包括错误的分子会导致模型精度显著下降。在过去,我们偶尔也会做出同样的观察。
在下文中,我们提供了两个例子,说明数据管理所需的高度努力,以及基于这一努力而取得的成功。几年前,拜耳与SIMULATIONS Plus合作,提供了19,500种化合物的实验pKa值,然后与公共来源的数据相结合。拜耳精心策划和注释的结构与最先进的机器学习相结合,使模型的平均绝对误差从0.72降至0.5个对数单位[54],从而成为表现最好的PKA模型之一[55]。
为了模拟药物化合物的代谢命运,并为I期和II期药物代谢创建代谢物位置(SOM)模型(稍后将介绍),我们严格管理了来自多个数据源的化学转换。在我们名为CypScore[56]的第一次尝试中,我们从文献中提取了大约2400个代谢转换。在我们的第二次尝试中,我们将这些数据与Accelrys代谢物数据库中经过整理和清理的信息结合在一起,产生了18,000个高质量的代谢反应,这些反应为细胞色素P450介导的代谢提供了质量提高,但还将适用范围扩展到非细胞色素P450[57]和II期酶[58]。
2.4机器学习算法
2.4.1有监督机器学习算法在药物发现中的历史
能够预测化学结构的标签或结果变量的化学信息学模型基于有监督的机器学习算法。这些有监督的学习算法的共同点是,它们将化学描述符作为输入,将分析数据作为期望的输出。Mitchell[59]和Lo等人[32]描述了当今药物发现中使用的ML算法的基本原理和局限性。此外,最近发表了一项非常系统的ML算法在化学健康和安全中的应用研究[60]。
在本世纪初,大约20年前,几个有监督的ML算法在化学信息学领域相互竞争。其中包括偏最小二乘(PLS)等线性模型和支持向量机(SVM)、人工神经网络(ANN)等非线性模型。线性模型已经流行了一段时间,因为它们能够预测物理化学参数,如依赖于增量化学碎片方法的logP[61],但也要归功于从Hammett和/或Taft方程得出的长期存在的线性自由能关系(QSAR)的概念。
然而,特别是生物化验数据的预测似乎更加复杂,而且并不总是线性相关。因为在许多生物检测中,一种或多种蛋白质(或其他生物分子)的识别起着作用,而这些结合过程不是递增的,相反,偶尔会突然不连续(所谓的活动悬崖),因此相关的生物学预测需要非线性算法也就不足为奇了。例如,当时在拜耳,对支持向量机、神经网络和k近邻(k-NN)等非线性监督算法进行了比较,以获得化学物质“类激酶”的最佳预测值[62],或者首选支持向量机来估计“类药物”[63]。
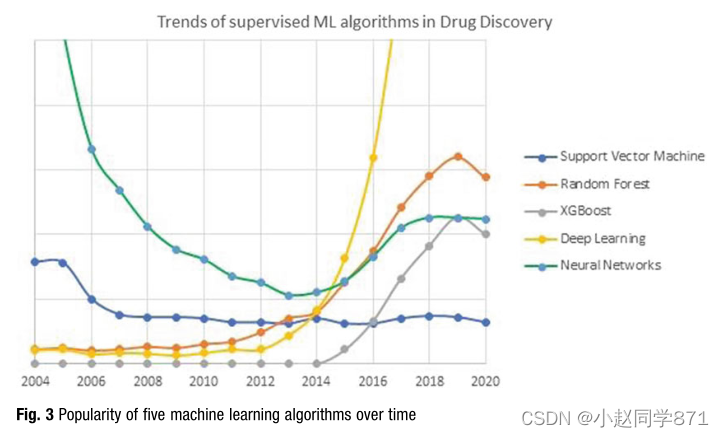
插图。五种机器学习算法随时间推移的流行程度
直到那时,决策树(DT)也被用作化学信息学中的非线性方法,然而,大多数成功是有限的,因为DT模型在工业应用中看起来一点也不健壮。在此过程中,基于Leo Breimann的工作,由树集合组成的随机森林(RF)似乎比决策树更健壮,在许多用例中具有总体良好的性能[64]。大约从2005年起,人们对随机森林的兴趣与日俱增,代价是其他非线性算法,如支持向量机和神经网络(见图3)。由于更强大的CPU,特别是GPU硬件的出现,神经网络的受欢迎程度从2015年开始缓慢下降,但又恢复了势头[65]。这使得神经网络(Deep NN)中具有更多隐藏层的更复杂的体系结构,与避免梯度消失的技术(如纠正线性单元(REU)或辍学等方法相结合,以避免神经网络出现的过度拟合问题,在2015至2020年间导致了深度学习的巨大突破[66]。
值得注意的是,在过去的二十年里,人们对随机森林(RF)的兴趣一直在稳步增长,尽管对深度学习兴趣的无与伦比的追赶。随机森林的稳定改进源于各种“树木增强”技术的发展,从1997年的Adabost开始[67],在定量构效关系中使用Friedman的随机梯度增强(SGB),即Svetnik等人[68],以及Chen和Guestrin的极端梯度增强(XGBoost)[69],因为这些作者能够最佳地结合现有梯度增强算法的各种特征。2014年,XGBoost出人意料地赢得了希格斯机器学习挑战赛。这一新算法后来被广泛使用,不仅是因为XGBoost的总体性能很好,还因为它似乎被设计成高效、灵活和可移植的。
2.4.2有监督的机器学习算法在药物研发行业中的利弊
值得注意的是,在过去的二十年中,与高效的现代非线性算法相比,线性最大似然算法,如偏最小二乘法或HQSAR(设计的描述符和偏最小二乘法的组合),偶尔在其性能上仍具有竞争力,尽管仅当期望的输出是一种增量性质的物理化学性质时,例如具有logP、logD、溶解度和膜亲和力的情况。同时,这些增量属性也在具有多任务设置的卷积神经网络中建模(见小标题2.9)[70]。
在用于监督最大似然的非线性算法中,随机森林长期以来一直是拜耳ADMET平台的选择方法,原因如下:(1)与环形指纹相结合,与神经网络、支持向量机和k-NN相比,RF模型的性能通常最高且非常稳健;(2)决定RF算法配置的开箱即用超参数通常是最优的,不需要像支持向量机那样进行搜索和优化。因此,RFS可以稳健地应用于自动再训练的预测平台,以及(3)随机森林等集合模型带来的“投票分数”可以用作个人预测的置信度度量。
深度学习的出现并没有自动取代随机森林在拜耳预测平台中的突出地位。在大多数药物发现引导优化活动中,分子的数量仍然适中(即<3000),DNN的性能并不优于RF来建立活性QSAR模型。在性能方面,如果数据集非常大,深度神经网络就会成为人们主要感兴趣的对象。相反,深度神经网络在药物发现中的优势似乎是:(1)不需要特征工程,这意味着可以在进行特征工程的同时学习特征(例如,图形卷积网络[70],(2)可以相当灵活地利用神经网络输入,这意味着如果数据是非结构化的或来源未知,甚至可以对模型进行训练。这意味着,即使数据的格式不同,也可以合并数据(即组合图像数据、光谱数据、活动数据等)。或者是否要以工业资产所有者保留对其信息的控制权和所有权的方式尊重专有数据的高度机密性,即MELLODDY(www.Mellowdy.eu)。
2.5描述符
尽管化学家对分子的看法通常是二维的,但现实是由核和电子组成的相互作用的、动态的、多构象的、灵活的实体。用于计算化学的分子的任何表示总是带有一些信息损失的抽象。对于化学信息学和机器学习中使用的表示法尤其如此。
2.5.1分子描述符
在大多数机器学习的场景中,化学结构和分子性质之间的相关性是被学习的,就像在溶解度或目标亲和力的情况下一样,因此抽象作为完整分子的描述符而得到。
对于描述符的分类有不同的方案。最根本的区别是分子描述符和原子描述符之间的区别,这一点将在本节后面讨论。此外,还区分了实验参数logP、熔点、摩尔折射率和从分子结构中得出的理论参数。此外,在实验参数,如logP,熔点,摩尔折射率,和从分子结构得到的理论参数之间。
这里我们将遵循维基百科(https://en.wikipedia.org/wiki/Molecular_descriptor),的分类方案,该方案定义了五个主要类别:
1.0D描述符(即构成描述符、计数描述符)、
2.1D描述符(即结构片段列表、指纹)、
3.2D描述符(即图形不变量)、
4.3D描述符(例如,对于3D-Morse描述符、突发事件描述符、逃逸描述符、量子化学描述符、大小、空间、表面和体积描述符)、
5.4D描述符(诸如从GRID或CoMFA方法、Volsurf得到的那些)。
由于Todeschini和Consonni的《分子描述符手册》[71]提供了自20世纪50年代以来派生的约1800个描述符的全面汇编,下面我们将重点介绍一些描述符类。
0维描述符,即分子性质,如分子质量或亲脂性,以及计数值,如给体、受体、环、卤素的数量,是直观的,但提供的信息不多。此外,它们高度相关,这进一步减少信息量,如表1所示,表1提供了随机选取1%的拜耳化合物甲板[72]的Lipinski[21]规则和Veber[25]属性的皮尔逊相关系数。
表1利平斯基(浅灰色)和维伯(深灰色)参数的特性相关性。Lipinski参数是受体原子数(#ACC)、给体原子数(#Don)、分子量(MW)和亲脂性(aLogP;在最初的出版物中是logP)。Veber参数是可旋转键的数目(#转键)、极性表面积(PSA)和供体和受体的数目之和(#DonAcc)。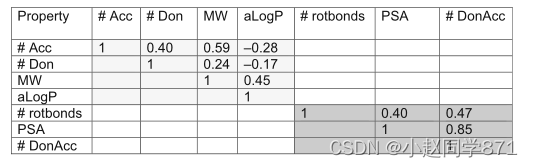
根据维基百科的说法,结构指纹是一维描述符(位或数字的矢量),它始于20世纪80年代,以Daylight指纹[73]作为快速查询化学结构数据库的手段开始使用。很快,事实证明,日光指纹及其近亲就像MACCS密钥(MACCS Structure Key)。2011年。Accelrys,San Diego,CA),它们是位向量,其中每个位指示预定义化学片段的存在或不存在,是机器学习的强大描述符[74,75]。
我们十多年来的主要描述符,被许多出版物证实,是圆形扩展连接性[76]指纹(ECFP),它将原子及其邻居的性质编码成一定拓扑(与起始原子的键数)半径和特征类型(元素、给体、受主等、原子类型)的位向量。描述符的长度为32位,编码42亿个不同的值。因此,它是非常稀疏的,通常折叠到1024或2048位。根据结构多样性和终点的不同,每项任务的最佳设置必须始终通过反复试验来确定(参看。第2.7节稳定模型和性能模型的识别)。
图不变的2D描述符,如拓扑或连通性指数,至少在我们手中通常会产生过度拟合的模型,这些模型在交叉验证中工作得很好,但不能对外部测试集进行预测。
基于分子的一个3D坐标集的3D描述符试图产生更真实的化学抽象。主要问题是它们对构象的依赖,这引入了歧义和噪音。分子的表面积、偶极矩、体积等性质可能有20%或更大的差异变形者。从GRID[77]或r CoMFA[78]方法得到的4D描述符还有一个额外的限制,即依赖于配体的排列,这有时不明显,只有在同类系列的情况下才可能。最后,有人试图克服构象依赖的问题,但受益于更真实的分子3D描述。形式上,根据维基百科的分类,基于多构象的可解释的无对齐xMAP[79]和基于多构象、互变构体和异构体的5D-QSAR[80]都是3D描述符,这说明这种分类有时是任意的。
尽管每个描述符都有优缺点,但据报道,每个描述符都有成功的申请[30,31]。现在有公共数据库和模型存储库,允许应用已发布的模型或浏览对复合集合的预测。例如:公共模型储存库QsarDB81、拥有200个模型和650,000种物质的丹麦(Q)SAR模型数据库、QSAR工具箱(https://qsartoolbox.org)),其中包括92,000种化学品的902个模型、57个数据库和2.6个实验数据点,或JRC QSAR模型数据库(欧盟委员会,联合研究中心(2020年):JRC QSAR模型数据库。欧洲委员会,联合研究中心(JRC)数据集,来自欧盟,记录提交给JRC的欧盟动物试验替代品参考实验室(EURL ECVAM)的模型的有效性。
预定义描述符的另一种选择是通过某种算法来学习分子的表示,该算法或者与用于学习属性的算法相关联,或者甚至是端到端地学习。实际上,直接从分子中学习最佳表示的看法并不完全正确,因为通常用作结构输入的微笑或因奇已经是分子的抽象表示(即简化)。2005年,维达尔等人以LINGO为首字母缩写提出了使用微笑进行机器学习的想法,后来数据科学界对其进行了重新发明。
表示学习始于Duvenaud等人将处理神经网络中的图的概念转移到分子[84]。在这里,原子对应于图中的节点,并结合到连接节点的边。特征是在节点级别学习的,使用图的邻接矩阵在相邻节点之间传递信息。像PotentialNet这样的类似方法也被开发出来[85],并被详细总结[86,87]。
不是从分子图中提取特征,而是可以使用变分自动编码器[88,89]来转换离散的对持续不断的陈述微笑。在学习阶段期间使用耦合到编码器的解码器来转换回微笑,并通过最小化重建误差来确保表示是适当的。序列到序列学习是Xu等人应用的另一种类似但无监督的方法[90]。
温特等人[91]应用自动编码器-自动解码器的概念来学习一组固定的连续数据驱动描述符cDDD,方法是在训练过程中将随机微笑转换为规范微笑。最终得到的描述符是基于来自锌和PubChem数据库的大约7200万种化合物。通过在8个QSAR数据集上的模型性能和在虚拟筛选中的应用,验证了该方法的有效性。它表现出与各种人工设计的描述符和图形卷积模型相似的性能。
2.5.2原子描述符
与原子的反应性有关的机器学习问题,如反应速度和区域选择性、pKa值、代谢命运的预测或氢键相互作用,需要将原子及其周围的性质编码到专门的原子描述符中。本书第[92]章的作者之一提供了关于原子描述符的全面概述。由于决定反应性的是原子上的电子分布及其化学嵌入,因此应用量子力学是描述符导出的明显选择。
在许多应用中,描述符值直接从量子化学计算[93]中检索,例如用于识别亲电芳香取代反应中的反应点的反应或过渡态能量[94,95],用于pKA估计的原子电荷[93][96]或基于概念密度泛函理论[97]的参数,例如用于共价结合的亲电性指数[98,99]。
对于其他应用,设计了由量子力学描述符组成的描述符,如基于构象、方法和基组不变原子电荷[100]的径向原子描述符,我们将其应用于新陈代谢部位(SoM)[57,58]、氢键强度[101,102]和Ames突变性[103]的预测。
但也有性能良好的经典邻域编码原子描述符的例子,用于SOM预测[104,105]和Diels-Alder反应的区域选择性[106]。
2.6性能指标
对模型进行适当的评估是至关重要的,因为对于生产性用途,它们需要准确和稳健,即在某一时间范围内稳定和可预测。嵌套交叉验证(CV)和独立测试集中的模型质量评估确保了在用于训练的化学空间之外的稳健性能。
不同的衡量标准不仅必须应用于不同的模型类别,如分类问题或回归问题,而且还取决于模型所解决的生物学问题。特别是对于回归模型,在生物相关值范围内的良好表现比总体表现对可能范围的影响更大。
回归模型的常用度量包括R2(R平方,不能与皮尔逊相关系数混为一谈)、均方根误差(RMSE)和Spearman‘s Rho。R2是决定系数,它提供了有关数据与回归线的接近程度的信息。完全关联将导致R2为1,但通常在0和1之间。理论上,R2可以是?1。如前所述,可能需要只计算预测属性的相关值范围的R2,而不是整个范围的R2。
RMSE是残差的标准差,指示预测值与实际数据点的接近程度,是可靠的通用误差度量。有时报告的类似度量是平均无符号误差(MUE)和平均绝对误差(MAE),但RMSE更可取,因为它更强调模型的偏差。
Spearman‘s Rho是一个非参数等级相关系数。对于根据实验值从低到高的预测的完美排序,Rho将是1。对于每一对错序的对象,它将减小,对于完美的逆排序将是?1。Rho的高值表明应用于项目中的模型将能够回答这个问题,例如,新的合成方案将比现有的分子更容易溶解。它不会回答再增加多少的问题。
用于评估分类模型质量的常用度量是从混淆矩阵(也称为关联矩阵)中推导出来的。混淆矩阵提供了真阳性、真阴性、假阳性和假阴性预测的数量。从这些数字中,可以得出多个度量。
总体精度是正确预测的对象在所有对象中所占的比例。在高度不平衡的数据集的情况下,准确性可能具有误导性,例如,如果模型总是预测较高的填充类别,则它将获得高精度而不具有预测性。在这里,平衡的准确性将是有用的,因为它是特异度和敏感度的算术平均值。
特异度或真阴性率,是被预测为观察到的阴性的比例,而敏感度,也被称为真阳性率或回忆,是被正确预测的观察到的阳性的比例。与特异性和敏感性相对的是假阳性和假阴性率,它们分别报告了错误预测的阴性/阳性在所有观察到的阴性或阳性中所占的比例。另一个更注重预测值而不是观测值的指标是正值预测值,也称为精度,它显示正确预测的积极在所有预测的积极中所占的比例。对于负面预测,这称为负面预测值。
关注正值的组合指标是F-Score,它是精度和灵敏度的调和平均值。最常用的F-Score是F1,精确度和敏感度是平均加权的。
马修斯相关系数(MCC)是回归系数的几何平均值,也适用于类别分布不平衡的分类问题。
最后但并非最不重要的是,科恩的kappa也是一个很好的衡量标准,它可以处理不平衡的类别分布,并表明与根据每个类别的频率随机猜测的分类器相比,该分类器的性能要好得多。
另一个流行的指标是接收器操作特征(ROC)图,用于可视化分类算法的性能。它描述了所有可能的分类阈值的真阳性率和假阳性率之间的相关性。理想的ROC曲线应该是从(0,0)到(0,1)到(1,1),既没有假阴性也没有假阳性预测,代表了一个完美的分类。从(0,0)到(1,1)的对角线表示无歧视的线,因此是最坏的情况。ROC曲线下面积(ROC AUC)是用于描述ROC曲线的数值度量。
2.7稳定模型和性能模型的识别
目前的审查[31,107]描述了被接受为机器学习最佳做法的过程,这一过程是在过去20年中发展起来的,现在普遍适用,并在经合组织各自的指南[108,109]中详细概述(见图4)。该指南是在制定欧盟关于化学品对人类健康和环境的潜在影响的REACH(化学品注册、评估、授权和限制)法规的过程中制定的。不遵循这些最佳实践通常会导致模型在其预期的应用场景中无法执行,这由Stouch等人描述[32]。
简而言之,主要包括以下几个步骤:
1.训练数据的准备,即化合物标准化和分析数据的预处理。
2.将数据集分成训练集、验证集和外部测试集。
3.描述符的计算。
4.算法的选择和相关超参数的优化。
5.模型培训,包括内部验证策略的应用。
6.使用适当的度量建立绩效评估模型。
7.基于内部验证步骤选择的模型的外部验证。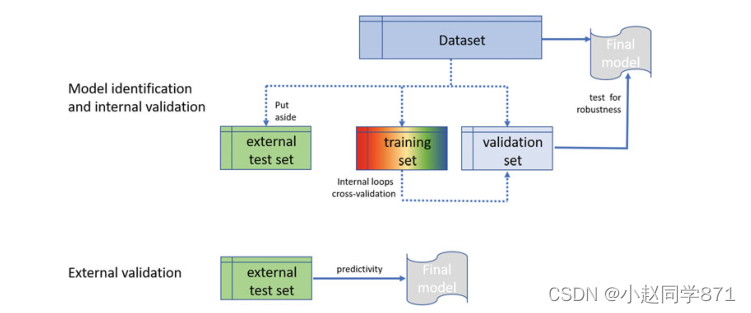
插图。识别稳定模型的一般程序,包括基于分为训练集、测试集和外部验证集的数据集的内部和外部验证。(改编自经合组织指南)
步骤3-6通常必须针对多个描述符集、算法和至少两个验证策略来执行。仅对于选择用于内部验证的模型,执行外部验证步骤作为对模型稳健性的最终检查。广泛应用的验证策略有交叉验证、自举和Y置乱。
交叉验证是通过取出对象的子集来执行的,然后由从其他对象建立的模型使用该子集进行预测。只有对于具有大约20-50个对象的非常小的数据集,留一法(L-O-O),即只留一个对象,是一种可以接受的策略。否则,较大的数据集通常被分成三到十个子集,例如,在五个子集的情况下,引导到五个子集中的四个上的每个模型,并预测第五个子集。通过多次执行此操作,就可以从每个对象的多个预测中得出一些性能统计信息。
相反,自举是重复的随机采样和替换,允许这些样本中的任何一个包含一些对象不止一次。Bootstrapping估计实验中的抽样分布,并识别预测误差和可信区间。将任何合适的算法组合成一个集合模型,然后给出一个集合预测,包括对预测不确定性的估计,这不太容易过拟合。随机森林就是这种建模技术的一个例子。
最后,Y置乱是一种技术,其中因变量在对象之间随机排列,作为对机会相关性的统计测试。如果可以确定性能良好且稳定的模型,还可以采取步骤8和9:
8.实施和应用最优模型。
9.模型再培训的设置和实施。
根据数据类型和数值分布的不同,并不总是可以得到一个数值模型。在这种情况下,必须参考分类模型,该模型通常(但不限于)报告两个类别,如活动和非活动。比较数字模型和分类模型的性能所需的不同指标将在单独的一节中介绍。
最后,由于数据集中涵盖的化学成分比潜在的化学成分少得多,所以总会有一些分子不在模型的适用范围之内。关于这一参数的各种尝试将在下一节中介绍,以及随着时间推移而降低绩效和再培训的主题。
2.8适用范围
通常,我们的监督ML模型基于有限的训练集,即数据要么来自项目中具有特定目标活性和受限化学空间的分子,要么来自不同研究项目的不同分子,因为它们在标准化的物理化学或药物动力学分析中进行测量。在这两种情况下,与巨大的类药物空间相比,模型预测可靠的化学空间区域是有限的,而且对完全新的分子的预测精度实际上可能令人失望。
这促使我们在我们的内部性质预测平台中向药物化学家提供更多信息:对于一些预测(即新陈代谢稳定性、Caco-2渗透和外流),预测的置信度(“可靠性”)旁边提供预测本身,以支持对单个计划或尚未测量的分子的判断(参见图5)。
近年来引入了许多不同的所谓适用域(AD)措施,这些措施可以分为两类。对未来物体在训练集中的嵌入程度进行距离测量的方法称为“新颖性检测”。量化到分类器决策边界的距离的方法称为“置信度估计”。尽管前者可以应用于使用例如到整个训练集的余弦、TAnimoto或Mahalanobis距离的任何算法,但后一种测量依赖于算法。最近,一项关于AD分类措施的广泛基准表明,信心指标总体上更好[110]。替代概念,如保角预测[111]或使用一种最大似然方法估计另一种方法的AD[112],仍需证明其有用性。
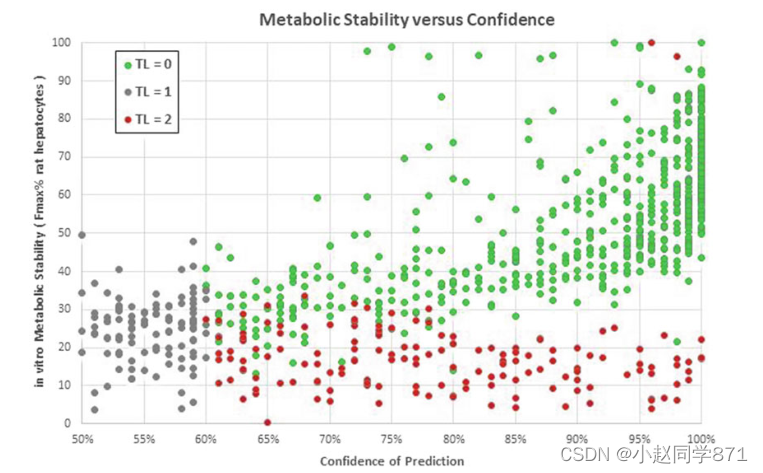
插图。举例说明类别概率估计如何区分可靠和不可靠的预测。使用拜耳每周自动训练的随机森林模型,对Menin-MLL Lead优化项目中的847种抑制剂的大鼠肝细胞体外代谢稳定性(FMAX%)进行了回溯预测[113]。随机森林模型预测类别标签(交通信号灯TL?2(红色)表示FMAX%<30%,交通信号灯TL?0(绿色)表示FMAX%>30%)和预测的可信度(选票分数)。如果单个预测的置信度降至60%以下,则预测平台将交通信号灯设置为TL≈1(灰色)以表示不可能有真实的预测
如上所述,随机森林在设计上是集合模型,因此带来了其内置的置信度估计器,即通过投票分数给出的类别概率估计。这种类型的置信度估计甚至可用于随机森林回归模型,而用于支持向量回归(SVR)的良好的可比较置信度度量仍然缺乏。
然而,支持向量机确实有一个令人满意但性能稍差的AD度量,用于基于Platt标度的分类。同样,神经网络提供了一种适当的AD度量,其性能优于随机森林,但质量高于k-NN和LDA[110]。
2.9适用于复杂和多个终端的型号
2.9.1基于多任务图卷积网络的物理化学ADMET终点建模
尽管上一节讨论了单任务模型,即每个属性一个模型来预测,但是最近的多任务建模[114]应用深度神经网络[115]来通过一个模型预测多个属性(端点)已经成为机器学习的许多领域的越来越先进的技术,并且这样的设置现在也被应用于药物发现任务[116-119]。
通过在所有任务之间共享(某些)隐层中的参数,多任务神经网络强制学习对所有任务有用的输入的联合表示。多任务学习的主要优势是(1)正则化,因为模型必须使用相同数量的参数来学习更多任务;(2)转移学习,通过与学习相关的任务帮助提取更一般方式有用的特征;以及(3)数据集扩充,通过将较小的任务与较大的任务相结合以避免对较小的任务过度适应[114]。
2016年,Kearnes等人[118]进行了第一项基准研究,比较了单任务(随机森林、Logistic回归)和多任务(深度神经网络)算法以及半径为2(类似于ECFP4)的1024位圆形指纹在22个顶点admet端点上的影响,包括Herg抑制、水溶解、化合物代谢和其他,约有28万个实验值和用作辅助信息的附加数据。虽然多任务模型的权重消除了数据集的不平衡,但单任务模型的深度神经网络得到了优化,而单任务基线模型使用了未优化的超参数,但总体结论是:多任务学习可以提供比单任务模型温和的好处,并表明较小的数据集往往比较大的数据集从多任务学习中受益更多。此外,我们发现,相对于更简单的多任务学习,添加大量的辅助信息并不能保证提高性能。我们的结果强调,多任务效应高度依赖于数据集,建议使用特定于数据集的模型来最大化整体性能。
然而,我们最近应用多任务图卷积网络,即表示学习和多任务模型的组合,预测了10个物理化学终点,这些终点决定了化合物的吸收和分布,在较小程度上决定了化合物的代谢和排泄[70]。
虽然我们已经有了在缓冲液pH值为2.3时和7.5时的logD[120]和膜亲和力(索维塞尔技术、https://www.sovicell.com)以及人血清白蛋白结合)的良好模型,但现有的在pH值为6.5时二甲基亚砜在PBS缓冲液中的溶解度回归模型不足以报告值,而是转移到分类输出。在那个时间点,我们也没有粉末溶解度和熔点的模型。最后,我们包括了另外三个溶解度数据集作为辅助任务,以丰富化学空间和信息内容。由于亲脂性、溶解性和熔点是相关的,这是众所周知的,我们希望通过结合这些任务来获得一些好处。
结果表明,多任务图卷积网络的性能与单任务图卷积网络相当或更好,优于单任务随机森林或具有圆形指纹描述符的神经网络,特别是在可解性的情况下,改进是突破的。
从这里给出的例子,以及其他一些出版物和我们过去几年的经验,我们可以说,多任务设置并不总是产生比单一任务在统计上显着的改进。它们也有缺点,比如模型培训的计算成本较高,以及一定程度上的过度适应风险。还有一个问题是,在具有自动再培训的环境中,优化的超参数随时间的稳定性。当我们决定让模型有效地实现时,我们总是会考虑这些参数。
2.9.2活体终端的建模
物理化学终点(如脂溶性或溶解性)和ADMET终点(如CYP抑制或肝脏清除)在高度标准化的物理化学或生化体外分析中进行测试。数据质量取决于前面讨论的参数,如分析分辨率或化合物纯度。由于活体生物体的可变性和更复杂的测试设置,体内终点包括额外的误差来源。
由于口服给药是最方便的给药途径,所有药理物质约80%的剂型都是口服的[18],因此识别通过肠道吸收良好的化合物是很重要的。口服生物利用度F被定义为可产生药理作用的口服剂量的范围[122],因此是药物发现中最重要的药代动力学特性之一。它被定义为口服(Po)和静脉(Iv)给药后剂量归一化暴露的比率。暴露量被确定为在通常24小时内采集的多个血浆样本的曲线下面积。药物发现阶段的相关物种是大鼠。人体数据最早可以在I期临床试验中获得。然而,最终的目标是为这两个物种建立模型。
生物利用度由吸收、新陈代谢、与血浆蛋白和组织的非特异性结合以及排泄的叠加过程决定。可在体外测试的相关化合物参数包括溶解度、亲脂性、pKa值、膜通透性、未结合分数或肝脏清除率。对于单一化合物,基于生理的药代动力学建模(http://www.open-systems-pharmacology.org,HTTPS://www.simulations-plus.com/software/gastroplus),)将物种的身体描述为相互作用的隔室的方法,允许根据体重、性别、疾病状态等附加参数描述不同器官中随时间变化的暴露情况,其质量可用于例如用于确定儿科剂量或风险(https://pubmed.ncbi.nlm.nih.。
GOV/32727574)。但这种方法依赖于体外参数或其部分计算模拟的可用性,本身并不适合高通量预测。
在最近的综述[123]中总结了使用结构描述符、结构描述符、实验值或其组合作为输入描述符的先前的机器学习尝试,并且概述也可以在我们的出版物[121]中找到。据报道,这些模型的准确度通常在70%左右,作者一直怀疑这些模型只对有限的化学空间具有预测性。
我们进行了一项彻底的研究,使用了大约1900个精心挑选的数据点,这些数据点来自两个一致的内部测试,用于确定po和iv暴露,以曲线下面积(Auc)表示,以及前面提到的六个相应的实验终点,这些终点仅适用于化合物的子集。
我们还对这些应用了In Silico模拟。我们建立了PBPK模型,这些模型由实验型、硅型参数或以微笑为结构描述符的混合方法建立。在这里,微笑首先由前身ML模型转换成伪PK参数,然后用于基于生理的建模。此外,我们还实现了一个带有圆形指纹的随机森林模型。
我们能够表明,静脉注射后的药物暴露可以用体外或电子预测终点作为输入的混合模型进行类似的很好的预测,数值预测的折叠变化误差(FCE)分别为2.28和2.08。对于口服给药,由于额外的吸收步骤,由于额外的吸收步骤,暴露的FCEs更高,与基于硅的模型相比,体外输入预测的FCEs分别为2.40和3.49。最终采取的方法是仅基于化学结构的口服生物利用度低的二元警报。在这里,我们获得了接近70%的准确率和精确度,无论是应用混合模型,将输出转换为二进制警报,还是使用随机森林分类器。该模型现在被用来选择相关的分子,从而避免了不必要的实验。
2.9.3药物代谢模型
所有生命系统都已发展出防御潜在有害外来物质的机制[124]。对于药物物质,这称为药物新陈代谢。哺乳动物的主要器官是肝脏,但新陈代谢也发生在肠道、肺和其他组织中。
任何口服药物在进入身体其他部位之前都会先通过肝脏。药物的生物转化可能会产生几个严重的后果。药物的高清除量降低了有效剂量,从而降低了疗效。药物代谢物可能是有毒的,它们可以抑制代谢酶本身或诱导其产生,所有这些都会导致药物疗效的不可预测的变化,药物与其他药物的相互作用,甚至耐药性。
这种情况由于性别差异、基因多态性、年龄、饮食、生活方式和许多其他因素而变得更加复杂。
代谢变化分两个阶段进行。在第一阶段,大多数细胞色素P450酶通过氧化和还原反应增加极性。在第二阶段,过多的酶,如UDP-葡萄糖苷转移酶、磺基转移酶或谷胱甘肽S-转移酶,将特定片段结合到第一阶段的代谢物上,以供肾脏排泄。
根据许多不同的酶反应,人们可能认为不可能估计化合物中代谢的速度、程度和不稳定的部位。然而,在过去的20年里,在鉴定SOMS的实验分析能力方面的高工作量和局限性导致了许多计算方法,应用了对接、分子动力学、量子化学计算和机器学习,包括或不包括蛋白质靶标信息。读者可参考Kirchmair等人[124]对实验和计算方法的全面概述。
原子在代谢反应方面的不稳定性是由它们的化学反应活性决定的,即各个原子的局部电子密度和空间可及性,这就需要原子描述符而不是分子描述符,以及对原子而不是分子的机器学习。只有通过量子力学才能充分描述原子上的局部电子密度(同样受到感兴趣原子的化学邻域的调制)。
在过去的二十年里,出现了多种解决SOM的ML方法。在拜耳,随着时间的推移,我们开发了两种方法。鉴于我们的第一个模型CypScore[56]使用空间和半经验描述符,如定域电子亲和力和电离势,仅限于预测细胞色素P450介导的反应,我们的第二次尝试基于精细的带有原子电荷的径向原子反应性描述符[100]和显著扩大的反应数据库评估了18个I期和II期代谢转化[57,58]。该模型允许通过对配体的化学修饰来设计出代谢负债,如示例性所示[124]。
2.10应用示例
2.10.1拜耳集成ADMET平台
我们现在已经描述了机器学习模型的先决条件、过程和资格。最后也是最重要的两个步骤是:
1.在一个易于使用的平台上使用户可以访问模型;
2.不断地与用户交流和培训用户。
随着我们的集成数据检索和分析平台PIX于2004年在拜耳推出,拜耳制药公司和后来的CropScience的任何研究人员都可以使用在计算化学中开发的模型[75]。如图6所示,多年来模型的组合及其质量不断增加。在此情况下,手动模型再培训的努力变得越来越令人望而却步,同时我们和其他人[125-127]发现,定期的模型再培训对积极项目的表现有积极影响,即使每个间隔只增加20-50个化合物。
因此,我们建立了一条全自动管道,每周从仓库检索所有化验的原始数据,执行前面描述的数据清理和聚合步骤,并填充数据湖。准备好机器学习的数据从那里获取,每周再训练模型,应用前面确定的优化模型设置。单元测试、日志记录和通过电子邮件向负责的计算化学家发送报告可确保数据的完整性和模型的稳定性。此外,关于特定端点及其关于数据问题的特性的知识被透明地记录在处理管道中,而在过去,此类信息有时会丢失。我们的机器学习工业工程方法释放了科学家的资源,使他们能够定期检查模型设置,并探索新的方法和新的端点。
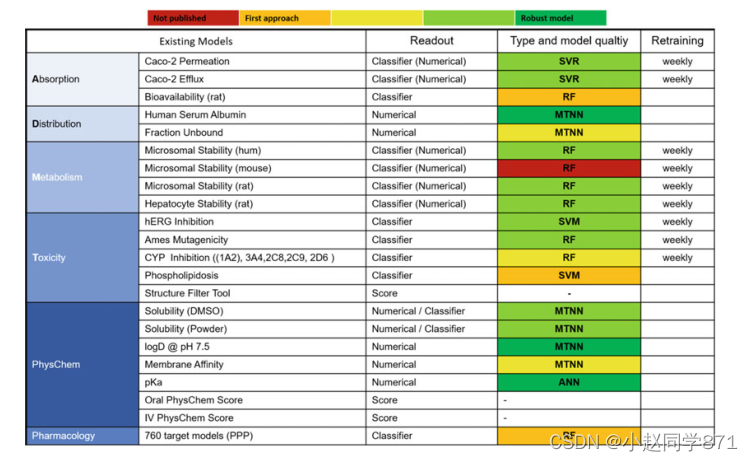
插图。拜耳的admet模型组合及其随时间的演变。通过颜色编码给出了模型性能的定性度量
这种新终点的选择是由项目需求和终点与决策的相关性驱动的,目的是优化药物发现过程。
2.10.2指导组合库的设计
与计算方法产生新的化学物质作为药物发现项目的起点,如虚拟筛选[128]或从头设计[129,130]平行,高通量筛选仍然是识别成功的有价值的工具。然而,实验测试对化学库有两方面的侵蚀,(1)物质消耗和(2)新颖性侵蚀,因为任何命中都会间接暴露化合物空间的某个子集。为了弥补这一点,拜耳最近开展了一项为期5年的活动–我们的下一代图书馆计划(NGLI),旨在通过500,000种新设计的化合物来增强HTS的收藏,正是为了应对这一挑战[72]。
我们选择结合两个领域的最好,因为实际的支架和合成计划是由药物化学家开发的,如果是基于结构的设计,则由目标类团队中的计算化学家通过“众包”进行,而最终的装饰是通过对多种化合物属性和多样性的帕累托最优化选择的,以实现每个化合物400-600个化合物的库。
2.10.3基于化学信息学和基于物理的方法相结合的线索优化
在一个典型的铅优化项目中,需要同时优化几个分子性质(例如,亲和力、效价、选择性、物理化学)。这些性质很少是相互独立的,例如渗透性的改善(admet性质)可能会导致溶解度降低(Physchem性质)。为了在多个优化参数中找到最佳折衷方案,必须进行复杂的多参数优化,这是药物发现项目中的一个关键挑战。有效地确定合成化合物的优先顺序是至关重要的,特别是当考虑到大的化学空间时。
在下面,我们概述了2016年的一个项目情况,在那里我们示范性地展示了如何通过结合化学信息学和基于物理的方法来解决来自大型虚拟化学空间的化合物的优先顺序问题。从之前针对相同蛋白质目标的优化运动中,我们有了一个我们手中的化合物具有高效力和良好的选择性,但在人类体内的半衰期不足。因此,目标是在保持领先化合物的高效性的同时改善PK性能。来自药物化学和计算分子设计的同事收集并列出了核心四个连接点中每一个潜在感兴趣的合成可及残基(图7)。
插图。举例说明给定分子核处残基的枚举的方案。核心区有4个替代位点和15个潜在的R1残基(R1:1。.15),9个R2的潜在残基(R2:1.。.9),R3的33个潜在残基(R3:1。.33),以及R4的14个潜在残基(R4:1.。.14)产生所列举的62,370个化合物的虚拟化学空间
列举所有可能的理想残留物组合创造了一个由60,000多种化合物组成的可合成的虚拟化学空间–在该项目的框架内合成这一数字是不可行的。不可避免地需要对这一巨大的虚拟化学空间进行有效的优先排序。我们专注于三个优化参数的挑战,以确定那些将显示出所需的低清晰度,低外排和高效的化合物(图8)。
如前所述,我们有一个强大的内部平台,包括定期接受再培训的基于ML的ADMET模型,以便快速确定优先级。由于可用的特定于项目的大鼠肝细胞清除量和Caco-2外排数据的数量和质量合理,我们建立了额外的局部模型。为了准确估计蛋白质与配体的结合亲和力,如果有适用的蛋白质结构信息,自由能微扰(FEP)等基于物理的结合亲和力计算是首选方法。基于高昂的计算成本,这种方法无法对60,000多种化合物进行效力排名。由于我们在所研究的化合物系列中观察到了高度添加的SAR,我们决定应用前面讨论的成熟的化学信息学Free-Wilson[131]QSAR(定量结构-活性关系)方法。经典的Free-Wilson方法将虚拟化合物的表征限制在那些包含已经合成的残基的新组合的化合物上。为了评估包括尚未合成残基在内的化合物组合的效力,我们用FEP计算的计算数据取代了缺失的实验亲和力数据(图9)。随后,我们将自由威尔逊QSAR方法应用于结合实验和计算的结合亲和力数据集。这种经典的化学信息学方法与基于物理的FEP方法的结合极大地扩展了传统的Free-Wilson方法的适用性。
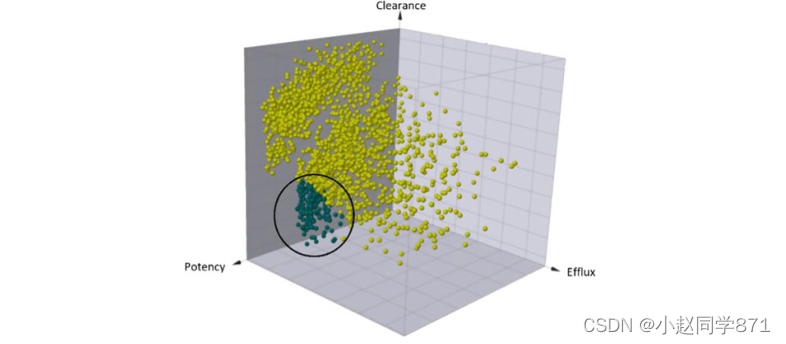
插图。定性地三维可视化了主要优化参数所跨越的空间清晰度、外排效率和效价。低间隙、低排出和高效力的所需空间中的虚拟化合物以深蓝色标记
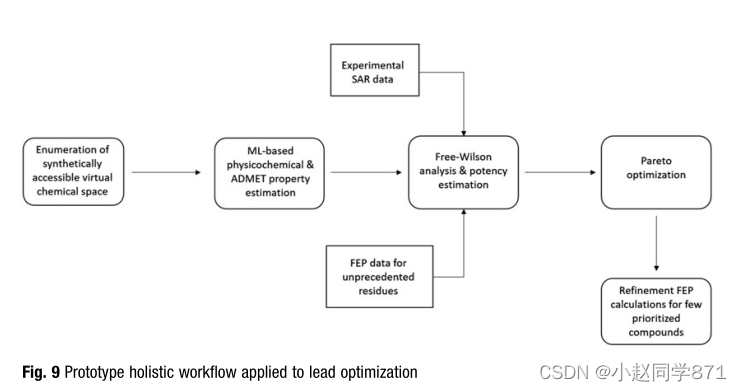
插图。原型整体工作流在线索优化中的应用
在一项旨在解决该方法准确性的概念验证研究中,我们使用薛定谔的FEP/REST(FEP+)方法计算了71个结合亲和力数据的最小集合[132,133],以基于Free-Wilson分析确定每个残基的效力贡献,并估计大型枚举虚拟空间中所有化合物的效力。
结合亲和力计算的准确性因目标和化合物类别不同而不同[133,134]。此外,应用方法、采样、系统设置、力场以及实验分析和计算数据之间的可比性是影响结果质量的关键因素[133-137]。对于最小的71种化合物中的28种,可以获得实验结合亲和力数据。我们观察到低平均无符号误差(MUE)为0.8千卡/摩尔。相应的实验测定的准确度可接近于0.2kcal/mol。对于来自大型虚拟化学空间的一组90个化合物,我们可以基于经典的Free-Wilson方法来估计其效力,并直接与实验进行比较。经典的Free-Wilson方法得到了低至0.4千卡/摩尔的MUE。使用纯粹基于计算的结合亲和力而不是实验的结合亲和力的Free-Wilson方法,我们观察到同一组化合物的Mue为1.6kcal/mol。1.6千卡/摩尔的MUE不足以满足铅优化的要求,但作为第一个过滤步骤仍然足够准确。
虽然这是一个不考虑过度匹配的原型例子(每个残留物只有一个样品),但它展示了确定大的虚拟化学空间的优先顺序的高潜力。为了获得更高的精确度,在过滤过程后的最后一步中,可以对少数选定的化合物进行显式FEP计算。帕累托优化使用我们在体外清除、外排和效力方面的主要优化参数实现了伴随的优先顺序。在此基础上,我们确定了从最初列举的化学空间合成的有前途的候选化合物。在该项目期间,不断涌现出关于核心可合成可获得残留物的新想法–导致化学空间不断增长。为了解释这种动态行为,整个过程的常规迭代变得必要,完全自动化在很大程度上是有益的。
目前,采用相关方法的自动化整体工作流程和平台正在演变[138],这些工作流程和平台将综合列举可综合利用的化学空间,并应用现有的基于ML和基于物理的模型,以确定化合物的有效优先次序。主动学习被逐步纳入,以提高模型精度,并反映Lead优化周期的迭代性质。最近的一个例子是在第一个过滤步骤中对物理化学性质进行多参数优化,然后在连续几轮中对接和应用在计算的FEP数据上训练的ML模型[139]。
在未来,我们预计会有越来越多的使用整体方法进行药物发现的成功应用实例[140]。
建模只是帮助指导药物发现过程的几个工具之一。后者仍然是一个充满挑战的过程,仍然以试错为主。建模在提供多少有用信息方面有其局限性,过去几十年来建模方法的发展既有成功也有失败。即使有强大的内部数据集和先进的技术,建模最终也不能消除对实验数据的需求,特别是在生物实验方面。
3.总结与展望
近年来,定量结构-活性关系(QSAR)[141]这一古老的概念引起了人们的极大兴趣。数据、算法和描述符是构建有用的基于数据的模型的主要组成部分,这些模型支持更高效的潜在客户发现和优化流程。这三个领域的重大进展是人们对这些模型重新产生兴趣的原因。关于数据,测试能力的自动化已导致整个行业的结构-活动数据显著增加。此外,愿意与区块链技术一起共享更多这些数据,使许多制药公司之间能够在保护隐私的情况下交换数据,使模型的数据基础增加了几个数量级。建模算法的改进,如(深度)神经网络和分子描述符,在许多情况下是为潜在的实验终点量身定做的,从而产生质量更高的模型。
在这篇综述中,我们讨论了ADMET数据的机器学习,因为这些数据是在许多项目的标准化可比分析中大量生成的,因为ADMET方面是反复出现的问题。其中一些机器学习模型已经达到了显著减少或停止实验测量的质量。然而,这并不是对所有端点都有效,尽管有大量的同质数据集可用,但一些ADMET端点仍然不能以足够的质量建模。对于药理学终点,数据是稀疏的,ML模型将只覆盖更大的多样化药物靶标组合中的一小部分(通常<30%),具有足够的预测性。因此,制药行业和生物技术领域的计算化学小组现在正致力于整体药物发现方法,将机器学习/人工智能和基于物理的建模相结合,以解决分子的效力/选择性、物理-化学/admet性质和可合成性。药理活性通常是解决了基于蛋白质结构的方法,包括对接/评分、自由能计算和/或机器学习模型。物理化学/ADMET的性质以及化学可合成性主要是用机器学习等基于数据的方法来模拟的。这种整体药物发现方法目前正在大量开发和测试中,可以预期,建模方法的智能组合将对未来如何进行小分子药物发现产生深远影响。

欢迎加入 MCP 技术社区!与志同道合者携手前行,一同解锁 MCP 技术的无限可能!
更多推荐

 1
1 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)